Chorea Huntington (erblicher Veitstanz)
Infobox
Diese Seite ist Teil einer Materialiensammlung zum Bildungsplan 2004: Grundlagen der Kompetenzorientierung. Bitte beachten Sie, dass der Bildungsplan fortgeschrieben wurde.
Infoblatt
Geschichtliches:
1872 beschrieb der englische Arzt Georg HUNTINGTON eine unheilbare Nervenkrankheit, die über Jahrzehnte hinweg Mitglieder jeder Generation einer Familie betroffen hatte und sich unter anderem in tanzartigen ( chorea = griech.: Tanz) Verrenkungen der Extremitäten äußerte.
Ursachen:
Chorea Huntington beruht auf der Mutation eines Gens (Genmutation) auf dem kurzen Arm des Chromosoms 4, einem Autosom.
Vererbungsschema (Stammbaum):
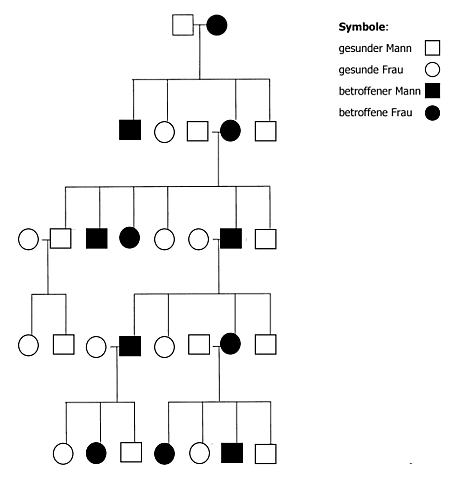
Abb. 7: Stammbaum einer von Chorea Huntington betroffenen Familie
Bildquelle: ZPG Biologie
Wie aus dem Stammbaum (Abb. 7) ersichtlich wird, überspringt die Krankheit keine der Generationen. Dies ist ein sicheres Zeichen für ein dominant vererbtes Merkmal. Da das Gen für Chorea Huntington auf einem Autosom liegt, wird der Vererbungsmechanismus als autosomal-dominant bezeichnet. Hierfür spricht auch das Auftreten der Krankheit unabhängig vom Geschlecht. Wichtig für das Verständnis von Stammbäumen ist, dass die Schwere der Erkrankung (Expressivität) bei einzelnen Erkrankungen und Erkrankten unterschiedlich ausfallen und somit auch falsch gedeutet werden kann. Die Durchsetzungsfähigkeit (Penetranz), ein Maß für die Wahrscheinlichkeit, mit der das dominante Allel zur Ausprägung kommt, kann bei unterschiedlichen Erkrankungen auch verschieden ausfallen. Bei Chorea Huntington beträgt sie 100%. Das heißt, alle Träger des mutierten Gens erkranken früher oder später.
Auswirkungen:
Die von der Krankheit Betroffenen sind zunächst vollkommen gesund und zeigen keinerlei Krankheitssymptome. Erst mit zunehmendem Alter (zwischen 30 und 60 Jahren) kommt es zum Ausbruch der Krankheit, die sich in Störungen der Gehirnfunktion, zunehmendem Gedächtnisschwund, unkontrollierbaren Bewegungen der Extremitäten und des Gesichts (Veitstanz) und Sprachstörungen äußert. Die Patienten werden mit zunehmender Ausprägung der Krankheit hilfloser und schließlich pflegebedürftig. Durchschnittlich 15 Jahre nach dem Auftreten erster Symptome endet die Krankheit mit dem Tod. Chorea Huntington ist deshalb besonders heimtückisch, weil die Betroffenen jahrelang frei von Symptomen sind und erst spät ihre Krankheit bemerken. Dann haben sie allerdings meist schon selbst Kinder, die mit 50%iger Wahrscheinlichkeit auch an der Krankheit erkranken werden. Neuerdings gibt es einen Test, mit dem mit 100%iger Wahrscheinlichkeit festgestellt werden kann, ob man Träger des mutierten Gens ist. Dieser Test ist bereits pränatal, d.h. bevor das Kind geboren wird, möglich. Eine Hoffnung auf Heilung des Defekts ist damit bis jetzt jedoch nicht verbunden. Damit ergeben sich natürlich vielfältige ethische Probleme, z.B. inwieweit sich ein Schwangerschaftsabbruch rechtfertigen lässt, oder ob man einem gesunden jungen Menschen erklärt, dass er später schwer erkranken und daran sterben wird.
Sonstige Krankheiten mit gleichem Erbgang:
Von 1500 bekannten monogenen (durch Mutation eines Gens verursacht) Erbkrankheiten werden 793 autosomal-dominant vererbt. Einige der bekanntesten davon sind:
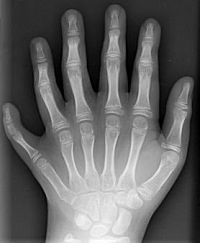
Abb. 8: Polydaktylie der linken Hand eines Kindes
Bildquelle: Polydactyly 01 Lhand AP.jpg von en:User:Drgnu23, subsequently altered by en:user:Grendelkhan, en:user: Raul654, and en:user:Solipsist. (Eigenes Werk) [GFDL oder CC-BY-SA-3.0], via Wikimedia Commons
Marfan-Syndrom (Bindegewebserkrankung, bei der durch ein einziges mutiertes Gen viele Merkmale verändert werden → Polyphänie.) Beim Marfan-Syndrom ist ein Gen für ein Eiweiß mutiert, welches zu erhöhter Elastizität des Bindegewebes führt. Deutliche Symptome dieser Erkrankung sind z.B. Hochwuchs, die Ausbildung einer Trichter- oder Kielbrust und öfters auch eine senkrechte Überentwicklung des Kopfes („Langschädel“). Die Expressivität des Marfan-Syndroms ist variabel, d.h. die phänotypische Ausprägung der Merkmale schwankt. In 20-40% der Fälle tritt das Marfan-Syndrom als Neumutation auf. Einer der prominentesten am Marfan-Syndrom erkrankten Personen war der amerikanische Präsident Abraham Lincoln.
Weitere Erkrankungen sind die Kurzfingrigkeit (Brachydaktylie), bei der eine Mutation zur Verkürzung einzelner oder mehrerer Finger führt, und die Vielfingrigkeit (Polydaktylie, Abb. 8), bei der es zur Ausbildung zusätzlicher Finger kommt.
Keine Krankheit, jedoch leicht für dich zu überprüfen, ist die ebenfalls autosomal-dominant vererbte Veranlagung, ob Urin nach dem Genuss von Spargel riecht oder nicht. Frage einmal in deiner Familie nach, ob das Phänomen bei deinen Eltern oder Geschwistern auftritt oder nicht.
Lernzirkel „Erbkrankheiten“: Herunterladen [doc] [1110 KB]
Lernzirkel „Erbkrankheiten“: Herunterladen [docx] [867 KB]
Lernzirkel „Erbkrankheiten“: Herunterladen [pdf] [553 KB]
Weiter zu Arbeitsblatt